
¿Qué es la detección de portadores de mutaciones de enfermedades recesivas?
La detección de portadores es una prueba genética que identifica mutaciones en genes relacionados con enfermedades recesivas, desarrollada para parejas en planificación familiar.
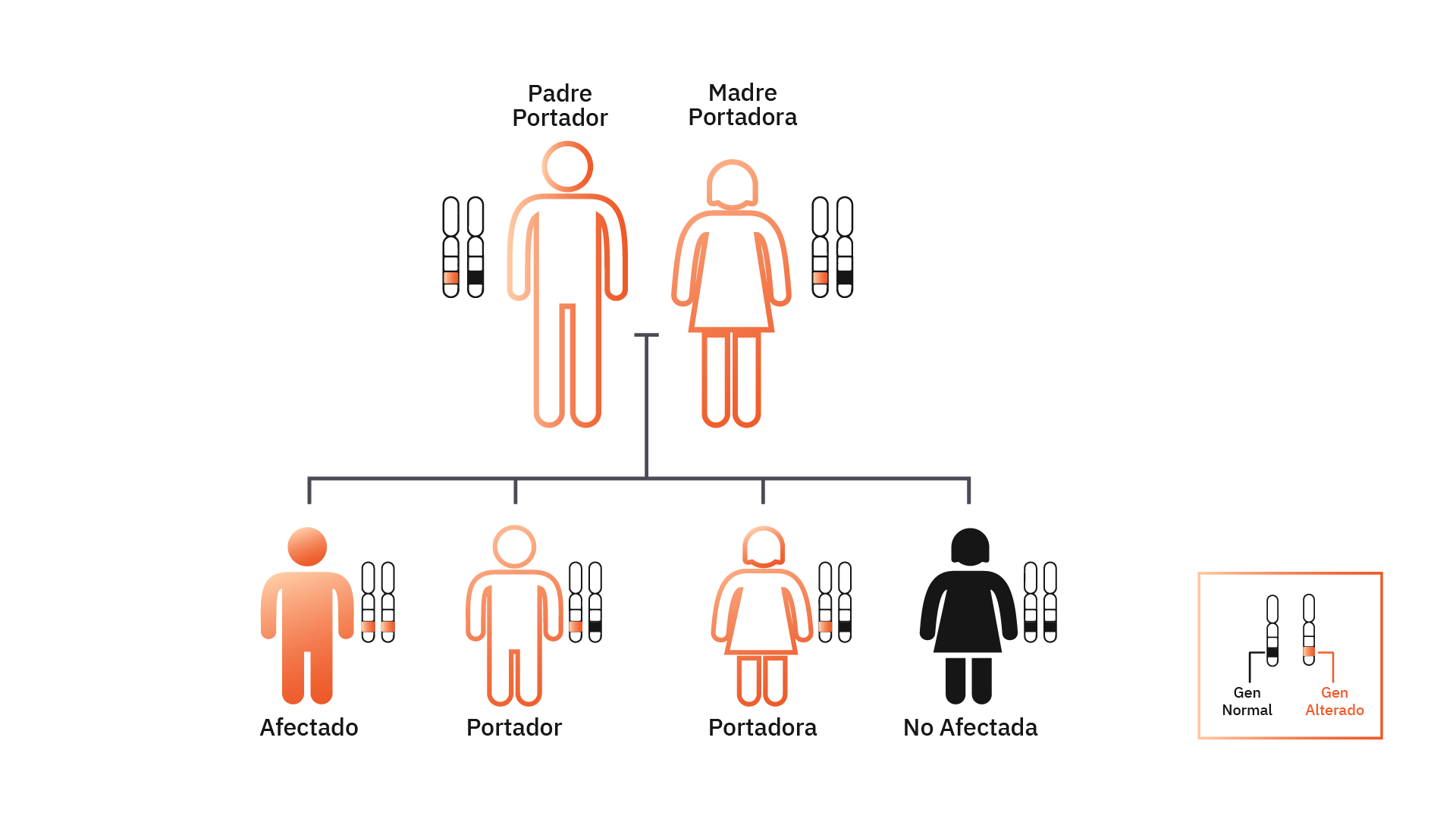
Comprenda cómo las enfermedades con un patrón de herencia recesivo se comparten en una familia.
¿La prueba debe ser ordenada por un médico?
La prueba de tamizaje no necesita ser solicitada por un médico, pero se recomienda que los resultados siempre sean monitoreados por un médico que asesorará a la pareja sobre las opciones de planificación familiar y los exámenes complementarios.
¿Qué esperar del resultado de este examen?
Si se hacen pruebas a ambos miembros de la pareja, el resultado puede proporcionar información sobre el riesgo de tener un hijo con una condición genética recesiva.
Si el resultado indica que la pareja tiene una mutación en el mismo gen, el riesgo de tener un hijo con enfermedad genética es al menos del 25%. En este caso, la pareja puede optar por la fecundación in vitro (FIV) y el diagnóstico genético preimplantacional (DGP).
¿Cuándo y quién puede realizar el examen?
- Parejas en planificación familiar
- Personas con mayor riesgo de enfermedades recesivas
- Clínicas de reproducción que trabajan con donantes de esperma y óvulos
Esta prueba debe usarse como una herramienta de asesoramiento genético para las personas con mayor riesgo de tener una mutación por enfermedades recesivas.
Indicado principalmente para familias formadas por parejas consanguíneas o para poblaciones con mayor riesgo de enfermedades recesivas, como la población judía, principalmente de herencia Ashkenazi, pero sin limitarse a ella, pudiendo extenderse a otras poblaciones como Sefaradim y Mizrahim, por ejemplo.
¿Por qué algunas parejas tienen mayor riesgo de tener hijos con trastornos genéticos?
Todos los genes humanos están en pares porque heredamos una copia materna y una paterna. La mayoría de las enfermedades genéticas son recesivas. Esto significa que para que la enfermedad se manifieste es necesario que las dos copias del gen (materna y paterna) estén mutadas (alteradas). Al heredar solo una copia de un gen mutado (copia materna o paterna), la persona solo será portadora de la mutación, pero no manifestará la enfermedad.
Cualquier pareja puede tener un hijo con una enfermedad genética, pero este riesgo es mayor para las parejas consanguíneas (son parientes), ciertos grupos étnicos con un mayor riesgo de enfermedades genéticas específicas (por ejemplo: judío asquenazí) o personas con antecedentes familiares de enfermedad genética recesiva como fibrosis quística y anemia de células falciformes.
Es más probable que estas parejas sean portadores de mutaciones en los mismos genes. Por eso es importante que las parejas en estas condiciones busquen especialistas para conocer sus riesgos antes de formar una familia y hoy en día ya existen pruebas genéticas capaces de ayudarlos.
Principales Enfermedades Analizadas
- Anemia de Fanconi C (FANCC)
- Deficiencia de dihidrolipoamida deshidrogenasa (DLD)
- Disautonomía familiar – Neuropatía sensorial y autónoma hereditaria (ELP1)
- Enfermedad de Canavan (ASPA)
- Enfermedad de Niemann-Pick A / B (SMPD1)
- Enfermedad de Tay-Sachs (HEXA)
- Enfermedad de jarabe de arce (BCKDHB)
- Fibrosis quística (CFTR)
- Glucogenosis 1A – Von Gierke (G6PC1)
- Hipoglucemia hiperinsulinémica 1 (ABCC8)
- Miopatía nemalínica 2 (NEB)
- Mucolipidosis IV (MCOLN1)
- Síndrome de Bloom (BLM)
- Síndrome de Gaucher (GBA)
- Síndrome de Usher 1D / 1F (PCDH15)
- Síndrome de Usher 3A (CLRN1)
Otras enfermedades analizadas en este examen
- Abetalipoproteinemia
- Acidemia glutárica I
- Acidemia isovalérica
- Acidemia propiónica
- Aciduria 3-metilglutacónica III
- Aciduria argininosuccínica
- Aciduria metilmalónica con homocistinuria
- Aciduria metilmalónica
- Agenesia del cuerpo calloso y neuropatía periférica
- Anemia de Fanconi A
- Anemia de células falciformes y talasemia
- Argininemia
- Artrogriposis, discapacidad intelectual y epilepsia
- Aspartilglucosaminuria
- Ataxia espástica
- Ataxia Telangiectasia
- Cistinosis nefropática
- Citrulinemia
- Deficiencia combinada de hormonas hipofisarias 2
- Deficiencia de 3-hidroxi-3-metilglutaril-CoA Liasa
- Deficiencia de acil-CoA deshidrogenasa de cadena corta
- Deficiencia de acil-CoA deshidrogenasa de cadena media
- Deficiencia de acil-CoA deshidrogenasa de cadena muy larga
- Deficiencia de biotinidasa
- Deficiencia de carbamoil fosfato sintetasa I
- Deficiencia de carnitina palmitoiltransferasa I
- Deficiencia del complejo III mitocondrial 1
- Deficiencia de factor XI
- Deficiencia de galactoquinasa (galactosemia)
- Deficiencia de holocarboxilasa sintetasa
- Deficiencia de lipasa ácida lisosomal
- Deficiencia múltiple de sulfatasa
- Deficiencia de piruvato carboxilasa
- Deficiencia de proteína trifuncional mitocondrial
- Deficiencia de vitamina E
- Deficiencia sistémica primaria de carnitina
- Disostosis espondilocostal 2
- Displasia esquelética
- Disqueratosis congénita 5
- Distrofia muscular congénita y anomalías cerebrales y oculares A3
- Distrofia muscular congénita y anomalías cerebrales y oculares A4
- Distrofia muscular congénita y anomalías cerebrales y oculares A5
- Distrofia muscular congénita, deficiencia de merosina 1A
- Distrofia muscular de cinturas 2A
- Distrofia muscular de cinturas 2B
- Distrofia muscular de cinturas 2C
- Distrofia muscular de cinturas 2D
- Distrofia muscular de cinturas 2E
- Distrofia muscular de cinturas 2F
- Trastorno de glicosilación congénita IB (CDG)
- Trastorno de glicosilación congénita IC(CDG)
- Trastorno de la biogénesis peroxisomal 1A / 1B
- Trastorno de la biogénesis peroxisomal 3A / 3B
- Trastorno de la biogénesis peroxisomal 4A / 4B
- Trastorno de la biogénesis peroxisomal 5A / 5B
- Trastorno de la biogénesis peroxisomal 6A / 6B
- Trastorno de biogénesis peroxisomal 9B
- Enfermedad infantil por almacenamiento de ácido siálico
- Enfermedad de Gaucher
- Enfermedad de Krabbe
- Enfermedad de Niemann-Pick C1
- Enfermedad de Niemann-Pick C2
- Enfermedad de Sandhoff
- Enfermedad de Segawa
- Enfermedad de Wilson
- Enfermedad del jarabe de arce
- Poliquistosis renal
- Encefalopatía por glicina
- Epidermólisis ampollosa de la unión
- Fenilcetonuria
- Galactosemia
- Gangliosidosis GM1 I
- Glucogenosis 1B / 1C
- Glucogenosis 2 (Pompe)
- Glucogenosis 3 (Forbes)
- Hiperfenilalaninemia por deficiencia de BH4
- Hiperoxaluria primaria I
- Hiperoxaluria primaria II
- Hiperoxaluria primaria III
- Hiperplasia suprarrenal congénita lipoide
- Hiperplasia suprarrenal congénita
- Hipofosfatasia
- Hipoglucemia hiperinsulinémica 2
- Homocistinuria
- Ictiosis congénita 1
- Inmunodeficiencia combinada grave (SCID)
- Intolerancia a la fructosa
- Leucodistrofia metacromática
- Leucoencefalopatía megalencefálica y quistes subcorticales 1
- Lipofuscinosis Ceroide (CLN) 1
- Lipofuscinosis Ceroide (CLN) 2
- Lipofuscinosis Ceroide (CLN) 3
- Lipofuscinosis Ceroide (CLN) 4A / 6
- Lipofuscinosis Ceroide (CLN) 5
- Lipofuscinosis Ceroide (CLN) 8
- Manosidosis lisosomal alfa B
- Miopatía de Nonaka
- Mucolipidosis IIA / IIB
- Mucolipidosis IIIC
- Mucopolisacaridosis 1H (Hurler-Scheie)
- Mucopolisacaridosis 3A (Sanfilippo)
- Mucopolisacaridosis 3B (Sanfilippo)
- Mucopolisacaridosis 3C (Sanfilippo)
- Osteopetrosis 1
- Paraplejia espástica (AAP) 15
- Picnodisostosis
- Retinosis pigmentaria 59
- Síndrome de Alport
- Síndrome de Alstrom
- Síndrome de Bardet-Biedl (BBS) 1
- Síndrome de Bardet-Biedl (BBS) 10
- Síndrome de Bardet-Biedl (BBS) 12
- Síndrome de Bardet-Biedl (BBS) 2
- Síndrome de Cockayne A
- Síndrome de Cockayne B
- Síndrome de Cohen
- Síndrome de Ehlers-Danlos VII
- Síndrome de Leigh
- Síndrome de Meckel 1
- Síndrome de Meckel 2
- Síndrome de Pendred
- Síndrome de Perrault 1
- Síndrome de ruptura de Nijmegen
- Síndrome de Sjogren-Larsson
- Síndrome de Usher 1
- Síndrome de Usher 1C
- Síndrome de Usher 2A
- Síndrome de Ellis-van Creveld
- Síndrome hidroletalus 1
- Síndrome nefrótico 1
- Síndrome nefrótico 2
- Síndrome de Neu-Laxova 1
- Síndrome poliendocrino autoinmune y displasia metafisaria reversible I
- Síndrome de Smith-Lemli-Opitz
- Sordera
- Tirosinemia I
- Tirosinemia II
- Trombocitopenia amegacariocítica congénita
- Xantomatosis cerebrotendinosa
- Xeroderma pigmentoso A
- Xeroderma pigmentoso C
Diferenciales de Mendelics

Pionero en Brasil y América Latina
Primer y más grande laboratorio enfocado en análisis genómico en América Latina.

Resultados más
precisos
Mayor base de datos de variantes brasileñas y latinoamericanas, con más de 100.000 exámenes realizados.

Calidad internacional certificada
Único laboratorio brasileño especializado en genética con CAP, PALC e ISO, principales certificaciones internacionales.

Agilidad en la entrega de resultados
Software de inteligencia artificial propio de Mendelics para analizar datos genéticos, ganador del premio MIT a la innovación.
